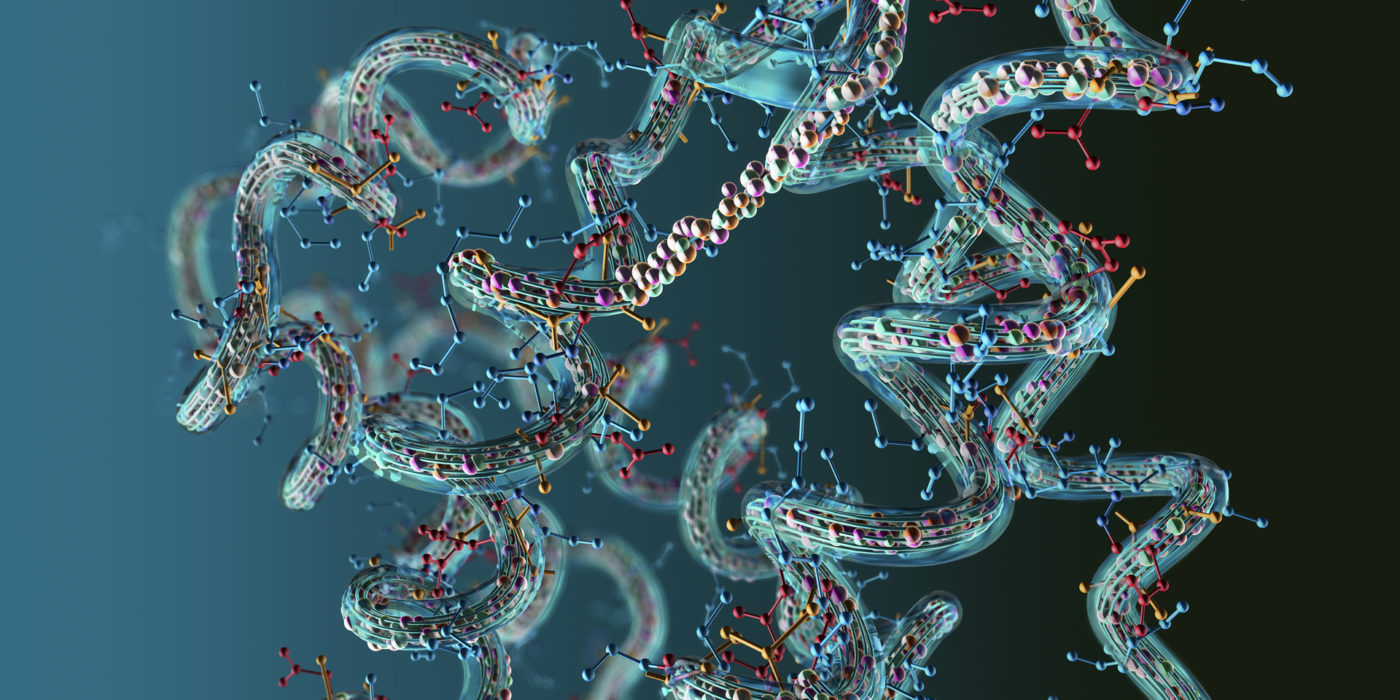
DeepMind выпустила третью версию программного обеспечения AlphaFold, что является значительным шагом в структурной биологии и искусственном интеллекте. AlphaFold3 на основе машинного обучения моделирует процесс сворачивания белков с высокой точностью, открывая возможности для лучшего понимания основных биологических процессов.
Сложность предсказания структуры белков
Белки представляют собой цепочки аминокислот, которые должны принимать точную трехмерную форму для выполнения своих биологических функций. Процесс складывания белков является основополагающим для жизни, поскольку определяет их взаимодействие с другими молекулами и выполнение задач, таких как катализ ферментов, транспорт молекул, клеточная сигнализация и иммунный ответ. Понимание трехмерной структуры белков необходимо для понимания их функционирования на молекулярном уровне.
Аминокислотная последовательность, или первичная структура, — это линейная цепь, которая складывается в сложную конфигурацию для приобретения функциональности. Сворачивание происходит под влиянием химических и физических сил: гидрофобных взаимодействий, водородных связей и сил Ван-дер-Ваальса. Клеточная среда с ее молекулами и условиями также влияет на процесс. Благодаря этому белки могут принимать разнообразные структуры. Незначительные изменения в последовательности аминокислот могут приводить к заметным различиям в конечной структуре.
Трудности предсказания с помощью моделирования
Сложность предсказания структуры белков по их первичной последовательности известна давно. Определение структуры белков традиционно осуществляется с помощью таких экспериментальных методов, как рентгеновская кристаллография, ядерный магнитный резонанс и электронная криомикроскопия. Эти методы даёт исчерпывающую информацию, но являются дорогостоящими, трудоемкими и ограничены конкретными типами белков.
Попыткой закрыть этот пробел стали вычислительные подходы к предсказанию структуры белков. Разработали такие методы, как моделирование гомологии, где структура белка определяется по сходству последовательностей с белками известной структуры, и моделирование молекулярной динамики, использующее принципы физики для моделирования сворачивания белков. Но эти методы имеют существенные ограничения по точности и масштабу. Развитие искусственного интеллекта открыло новые возможности в этой области.
Сила AlphaFold3
Разработанный компанией DeepMind программный пакет AlphaFold предназначен для моделирования сворачивания белков. Новая версия (AlphaFold3) может предсказывать не только структуру белков, но и их взаимодействие с биологическими молекулами, например ДНК, РНК и лигандами.
Молекулы-лиганды специфически связываются с белком-мишенью, создавая комплекс лиганд-белок. Молекулы могут быть различной химической природы: от мелких органических соединений до ионов металлов и макромолекул, таких как полисахариды. В структурной биологии лиганды часто выступают регуляторными молекулами или биологическими медиаторами, взаимодействующими с белками для управления их активностью. В фармакологии лиганды – это химические соединения, предназначенные для связывания с конкретными целевыми белками, например, клеточными рецепторами, для изменения их функции и лечения заболеваний.
Усовершенствованная программа AlphaFold, оцениваемая как на 50 % точнее существующих программных методов предсказания структуры белков и их взаимодействий, открывает новые возможности в медицине, сельском хозяйстве и биотехнологиях.
Главное ограничение заключается в том, что AlphaFold3, в отличие от предшественников, не имеет открытого исходного кода. Это значит, исследователи не имеют доступа к его коду и обучающим данным. Такое положение может затруднить научное сообщество при настройке модели или использовании ее для конкретных исследований.
Некоммерческие исследователи могут использовать сервер AlphaFold от DeepMind для получения предсказаний структуры белка на основе молекулярных последовательностей за короткий промежуток времени. В день доступно не более двадцати задач, что может быть недостаточно для интенсивного использования в некоторых исследованиях.