
Белок называют кирпичиком жизни, но это не просто расходный материал, а «строитель», «прораб» и «архитектор» в одном лице, обладающий тысячами специализаций и выполнение несметного количества функций. В организме человека около 20 тысяч генов, практически все кодируют белки, каждый из которых состоит из уникальной последовательности аминокислот. О вычислительном дизайне белков, их эволюции и процессе самопроизвольного сворачивания – наш разговор с профессором Сколтеха Дмитрием Иванковым.
Справка: Дмитрий Николаевич Иванков ― Кандидат физико-математических наук, биоинформатик, старший преподаватель и профессор Центра молекулярной и клеточной биологии Сколковского института науки и технологий. Под руководством Д.Н. Иванкова группа занимается изучением эволюции белков, их структуры, сворачивания и дизайна с использованием анализа данных, разработки алгоритмов, машинного обучения и молекулярного моделирования.
В 2024 году Нобелевскую премию по химии присудили за предсказание трёхмерной структуры белков, то есть расположения их атомов в пространстве, а также за разработку вычислительных методов проектирования белков. Как развивалась эта область исследований?
Можем считать началом 1961 год, когда Кристиан Анфинсен провел эксперимент, за который получил Нобелевскую премию по химии в 1972 году. В ходе эксперимента он доказал, что трехмерная структура белка определяется исключительно его аминокислотной последовательностью в организме. На основе этого открытия возникло предположение: зная последовательность, мы сможем быстро и эффективно предсказывать рабочую трехмерную структуру белков. С тех пор было предложено несколько подходов к решению этой задачи, но ни один не давал стопроцентной точности в предсказаниях. И лишь спустя более полувека, в 2020 году, в этой области случился долгожданный прорыв: ученые из компании… Google DeepMind Создали второй вариант известной программы. AlphaFoldСистема, которая достигла точности предсказания трехмерной структуры белка, близкой к результатам экспериментов.
― Около 90%?
Увеличение показателя с 50% до почти 90% оказалось существенным прорывом в данной научной области. Премия Нобеля за эту разработку — вопрос времени.
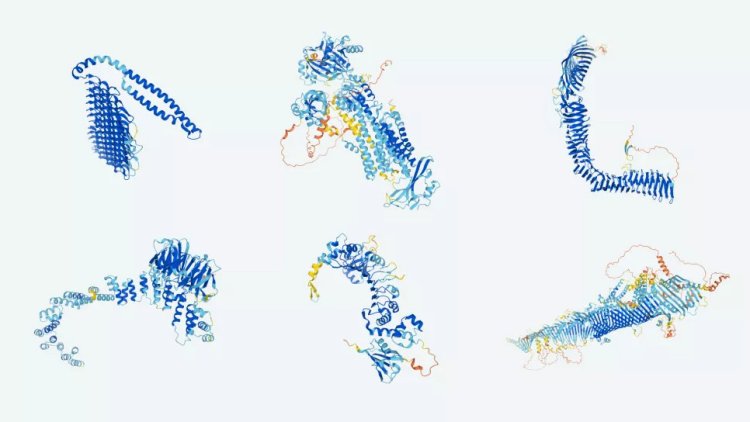
Программа AlphaFold определила трёхмерную структуру практически всех из двухсот миллионов известных науке белков. На фото: изображение белковых структур, предсказанных программой AlphaFold.
Фото: Google DeepMind
Ещё одна часть Нобелевской премии по химии досталась за создание белков с помощью вычислений.
Над задачей вычисления на компьютере белка с не встречавшейся в природе аминокислотной последовательностью ученые трудились с конца 1980–1990-х годов. Реальных успехов начали добиваться к концу 1990-х и началу 2000-х, прежде всего в группе под руководством Дэвида Бейкера.
Похоже, это как генерация изображений с помощью нейросети, только вместо картинок получаются белки.
Эта аналогия верна. Если вам интересна тема искусственного интеллекта, то вы вероятно знаете, что сначала его тренируют на изображениях кошек и собак, а затем отточенные технологии применяются в других сферах.
Зачем генерировать белки, не встречающиеся в природе?
Это пригодится в разных областях, например, при проведении реакций с белком-катализатором. В ожидании завершения химической реакции естественным путём могло бы пройти большое количество времени.
Прогресс в белковом дизайне позволяет создавать новые белки, которые могут существенно ускорять реакции и сокращать время получения конечного продукта.
Белки могут применяться для разрушения пластмассы – лишь один пример среди многочисленных перспектив.
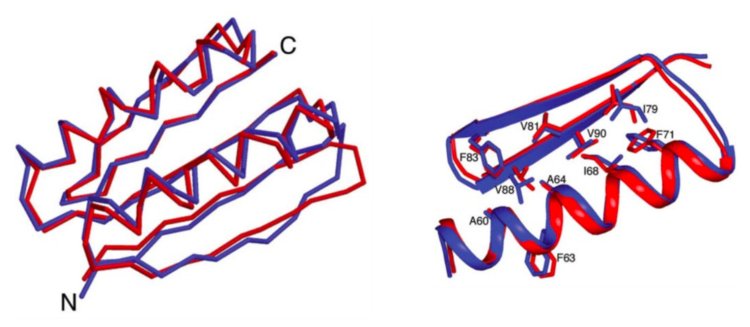
Топ7 — первый образец белкового проектирования.
Фото: Брайан Ку hlман, Гаутам Дантас, Грегори К. Ирэттон, Габриэле Варани, Бэри Л. Стоддарт, Дэвид Бейкер. / Science
Как полно понят механизм формирование белковой структуры тремя измерениями?
Довольно неплохо. Исследования начали в начале 1950-х годов. Первым белком с определенной аминокислотной последовательностью стал инсулин, а первыми с трехмерной структурой — миоглобин и гемоглобин. Главным вопросом стало сворачивание белков: как белок определяет свою конкретную структуру? Считалось, что рабочая структура наиболее стабильная. Но как белок ее находит? Вопрос поставил американский молекулярный биолог и программист Сайрус Левинталь, также один из первых, кто визуализировал структуру белка на экране осциллографа.
Сайрус Левинтал полагает, что белкам недостаточно времени существования Вселенной, чтобы изучить все доступные ей конфигурации.
Если он не изучит все варианты построения, предполагал Левинталь, то не сможет обнаружить самую устойчивую форму и принять её. Как же это работает? Проблема получила название «парадокс Левинталя», и многие учёные искали ей решение.
Левинталь высказал предположение о том, что белок складывается не в самую устойчивую форму, а в быстро доступную конфигурацию, выбранную эволюцией. В 1997 году Азат Бадретдинов и Алексей Финкельштейн доказали, что к самой стабильной структуре существуют быстрые пути сворачивания, а параллельные пути лишь ускоряют процесс.
Важно понимать, что с белковыми структурами это происходит не потому, что эволюция постаралась, а потому, что это свойство белковой глобулы как таковой. Среди множества путей сворачивания молекулы выбирают наиболее быстрый путь. Представьте работающий и сломанный эскалаторы. Вы выберете тот, что сможет доставить вас из точки А в точку Б быстро и с минимальными усилиями. Так же и здесь. Только «эскалаторов» в данном случае намного больше, чем два. Наличие сломанного эскалатора всё же увеличивает число людей, доставленных из точки А в точку Б.
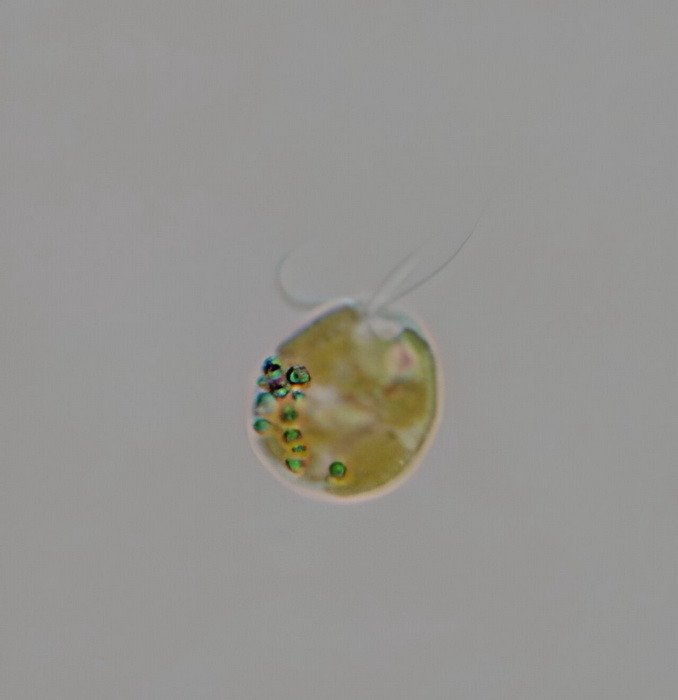
В 2024 году в клетках водоросли Prymnesium parvum был обнаружен наибольший известный белок — PKZILLA-1. Масса его молекулы достигает 4,7 МДа, а последовательность содержит более 45 тысяч аминокислотных остатков.
Фото: Greg Southard / Texas Parks and Wildlife Department
Парадокс Левинталя разрешили в 1997 году, и мы поняли, как сворачивается белок, но предсказать его структуру это не позволяло. Затем появились попытки смоделировать жизнь белка на компьютере, посмотреть, как он свернется, но скорость работы компьютеров недостаточна, чтобы сравниться с темпами протекания подобных процессов в природе. Долгое время нужно было ждать, пока белок свернется виртуальным способом. В 2010 году появились первые работы, где удалось свернуть очень маленькие белки, что можно считать прогрессом в этой области.
Внешние факторы воздействуют на данный процесс, или всё определяется исключительно внутренними причинами?
Внешние условия влияют на то, будет ли белок принимать трехмерную структуру или оставаться развернутым. Исключением могут быть случаи, когда у одного белка может быть несколько конечных конфигураций, и часть молекул сворачивается в одну форму, а часть — в другую, при этом переключение между ними часто обусловлено внешними факторами. Таких белков немного: около сотни на сегодняшний день.
Жак Моно, один из основателей молекулярной биологии, утверждал в своей книге «Случайность и необходимость», что возникновение жизни обусловлено способностью белков узнавать другие молекулы, включая другие белки, по форме. Что вы думаете об этом?
Это мысль интересна, и можно привести примеры распознавания такого рода. В природе множество белков, все разные. Рассматривая ферменты, например алкогольдегидрогеназу (белок, расщепляющий этиловый спирт), при благоприятных условиях он сворачивается сам по себе. Внешние условия определяют лишь то, будет ли он сворачиваться или оставаться развернутым. Для формирования структуры этого белка не требуются другие молекулы, которые он должен узнавать. Но есть и другие белки — нативно развернутые. Они находятся в развернутом состоянии до тех пор, пока не появится партнер, способный стабилизировать их структуру. Если такой партнер найдется, то взаимодействуя с ним, им выгоднее свернуться.
― То есть они распознают его, а затем сворачиваются?
Можно сказать так. К такому классу белков относятся, например, почти все рибосомальные белки. При наличии рибосомы белки сворачиваются на ней, поскольку взаимодействие с рибосомой делает их структуру рабочей и стабилизирует ее. В отсутствие этих взаимодействий белки предпочитают находиться в развернутом виде.
Возвращаясь к цитате Жака Моно, пример показывает класс белков, которые без партнеров предпочтительно остаются развернутыми, но при появлении партнеров распознают их и предпочитают находиться в свернутом виде в комплексе с ними.
Сколько таких белков существует? Что известно о их взаимодействующих молекулах?
Нет, это не единичные случаи. Зависит от того, кого считать партнерами. Если относить к ним молекулы воды, то получится, что такой партнер нужен абсолютно всем белкам для нахождения своей трехмерной структуры. Можно посмотреть на это с другой стороны: считать воду не партнером, а просто растворителем и сконцентрироваться на более специфических вещах, таких как взаимодействие белков с ионами металлов. Структуры некоторых белков стабилизируются взаимодействием с ионами цинка, железа и др. В данном случае именно ионы выступают в роли партнеров.
Существуют белки, трёхмерная структура которых стабилизируется благодаря сложным молекулярным образованиям, таковыми являются, например, гемоглобин и миоглобин, чья структура стабилизирована гемом.
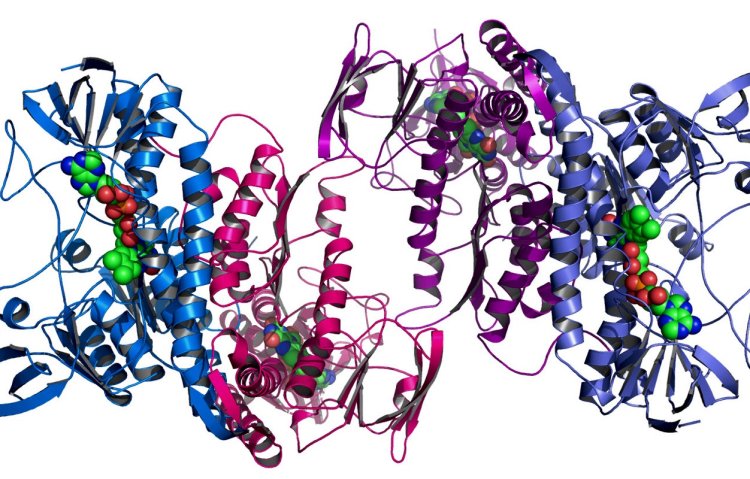
Длина цепи белков обычно варьируется от 200 до 400 аминокислот (размер может отличаться у разных организмов). На изображении – фермент из бактерии Colwellia psychrerythraea.
Фото: Andrzej Joachimiak / Argonne National Laboratory / Flickr
В теле человека сколько белков?
Тысячи белков выполняют практически всю работу в живых организмах. Их работа заключается во взаимодействии с другими атомными и молекулярными образованиями, часто — с нуклеиновыми кислотами. В человеческом организме около 20 тысяч генов, кодирующих разные белки. Таким образом, в нашем организме существуют 20 тысяч типов белков. Каждый белок обладает уникальной аминокислотной последовательностью и сворачивается в трехмерную структуру с жесткой формой, что позволяет выполнять предназначенную ему работу. Любой живой организм представляет собой огромный конвейер с тысячами работников-белков, осуществляющих все химические реакции и другие жизненно важные процессы.
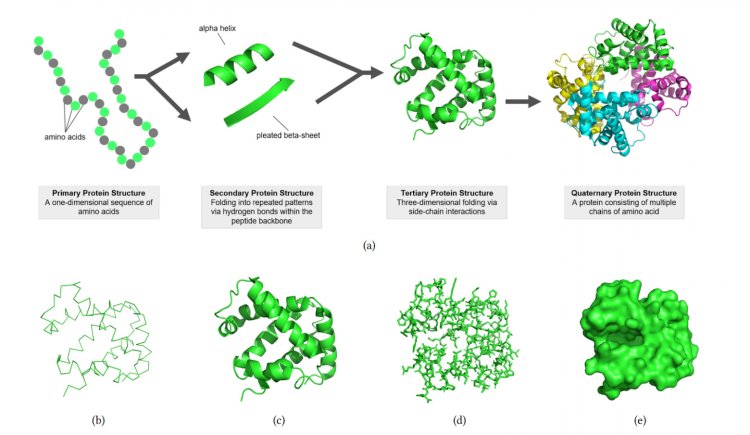
Последовательность аминокислот образует структуру белка, которая определяет его функцию.
Какие исследования по теме эволюции белков ведутся в вашей лаборатории?
Конечно. Сейчас сосредоточены на двух направлениях: исследовании стабильности белков и изучении их структуры и эволюции. Если говорить о первой теме, то хотелось бы, чтобы была разработана компьютерная программа, в которую можно было бы внести информацию о необходимых функциях белка, а затем получить на выходе аминокислотную последовательность такого белка для создания его в лаборатории. Это перспектива на будущее. Исследования в области компьютерного дизайна белков начались с самой простой задачи: изучения того, как предсказать изменение стабильности белка вследствие замены одной аминокислоты в его аминокислотной последовательности. Как ни странно, метода, предсказывающего результат таких аминокислотных замен, до сих пор не существует.
― С помощью программы Rosetta Нельзя ли так поступить и с Дэвидом Бейкером?
Да. Эта программа уступает другим. Мы изучали, как программы делают предсказания о влиянии мутаций на стабильность белков. Результаты показывают корреляцию с экспериментальными данными от 35% до 60%, но это недостаточно. Только при точности на уровне 90% можно будет считать задачу решенной.
― Как именно вы пытаетесь решить ее в лаборатории?
Разными путями. К примеру, создали собственную программу, предсказывающую изменение стабильности белка при мутации. Тренировались на новых данных 2023 года от Котаро Цубоямы и коллег: коллеги опубликовали замечательный… экспериментВ эксперименте, проведенном за неделю в одной лаборатории, изучили устойчивость 850 тысяч различных белковых вариаций.
― Это много?
Благодаря новым данным 2023 года удалось создать программу, демонстрирующую корреляцию с экспериментом на уровне 75%. Это значительно больше, чем было раньше: около 13 тысяч белков, определённых лабораториями мира начиная с 1970-х годов. Работа продолжается, разработчики программы стремятся к дальнейшим достижениям. ABYSSAL В расчётах применялись нейросети, методы молекулярной динамики и квантовой химии.
Различные представления структуры белка.
Фото: Луиз Филипе Веччиетти, Минджи Ли, Бегенч Хагенглиев и др. ArXiv.org
― Программой AlphaFoldТехнология, которую обсуждали ранее, применяют учёные во всём мире. Как это повлияло на ваши исследования?
Релиз программы был для нас очень значимым событием. Многих убеждают, что программа разрешила все проблемы сворачивания белка. Но это заблуждение. Процесс представляет собой множество сложнейших задач, а предсказание трехмерной белковой структуры – важное, но не единственное направление в этой области.
В 2022 г. мы первыми в мире опубликовали исследование, касающееся применимости структур, предсказанных AlphaFold, Программ для предсказания влияния мутаций на стабильность белков.
Мы стремились установить, достаточно ли… AlphaFold Для точности предсказаний структуры белков используются программы, которые позволяют нам опираться на рассчитанные ими модели вместо экспериментально определенных структур, так как таковые не существуют для всех белков. В нашей работе продемонстрировано, что для качественных программ, прогнозирующих изменение стабильности белка в связи с мутацией по его трёхмерной структуре, можно использовать модели, полученные AlphaFold, и потери точности не будет.
— Что вас больше всего привлекает в исследовании эволюции белков?
Нам очень интересно изучение эволюции как явления в целом, а не эволюция отдельных белков. Один из важных вопросов — это эпистаз. Понять его можно на примере любого функционального белка.
У вас есть белок, который эволюционирует: в нём происходят мутации, которые сохраняются, например, заменой одной аминокислоты на другую в аминокислотной последовательности. Из-за таких изменений в различных организмах встречаются варианты одного и того же белка, отличающиеся по последовательности и даже структуре. Но если эффект такой замены универсален и не зависит от предыдущих изменений в этом белке, то мы получаем полную предсказуемость! В таком случае можно было бы экспериментально измерить все эффекты всех одиночных замещений, а затем для предсказания эффекта любого количества замещений просто сложить эффекты этих одиночных замещений.
― Но этого не происходит.
Полная предсказуемость фенотипа по генотипу возможна только при отсутствии эпистаза. На самом деле эффект одной замены может зависеть от ранее произведенных в белке заменах. Эпистаз — сложное явление, изучение которого необходимо для прогнозирования свойств белка.
По моему мнению, вершиной эволюционной биологии могло бы стать появление программы, которая при загрузке аминокислотной последовательности белка выдаст информацию о его роли в организме: функции и необходимые для взаимодействия партнёры.
Какое максимальное число аминокислотных заменов может присутствовать в одной белковой цепи?
Эта тема очень интересна для обсуждения. Случайные замены приводят к тому, что примерно пять-шесть модификаций одного белка из 200 аминокислот дают 50% вероятности его неработоспособности. Если же делать точные замены на аминокислоты, встречающиеся у этого же белка в других организмах, то можно изменить до 70% остатков и белок продолжит функционировать.
Можно ли поменять аминокислоты у белков млекопитающих на рептильные, сохранив при этом работоспособность белков?
Говоря о белках, заметим: при сравнении одного и того же белка у людей, рептилий и грибов обнаруживается, что 70% аминокислотных остатков в этих протеинах различны. Несмотря на это, один и тот же белок выполняет одну и ту же функцию как у нас, так и у них. Удобные для изучения эволюции одиночные замены не единственный тип изменений: возможна потеря целого куска гена или его вставка.
― Это тоже мутации?
Да, это мутации, называемые делециями и вставками. При копировании генетической информации во время деления клеток фермент, отвечающий за копирование, может допускать ошибки.
― Не обусловленные воздействием внешней среды.
Наш организм подобен конвейеру, где белки выполняют роль работников. Каждый этап конвейера обслуживает определённый белок, работающий вместе с коллегами. Как и любой рабочий, белок может допускать ошибки. За жизнь человека в организме возникают от 20 до 100 новых мутаций, передающихся потомкам.
Для лучшего понимания этого процесса нужно изучить упомянутый вами эпистаз.
Мы разрабатываем алгоритмы для изучения правил эпистаза в нашей лаборатории. Стремимся понять, как замена одного аминокислотного остатка в белковой последовательности может повлиять на замену в других позициях белка. Поиск ответов на этот вопрос приблизит нас к возможности предсказания фенотипа по генотипу.